Retinitis pigmentosa:
Auf dieser Seite: Was ist die Retinitis Pigmentosa, wie wird sie festgestellt, gibt es verschiedene Formen und was kann man tun.
Definition:
Die Retinitis pigmentosa ist eine vererbbare, d.h. genetisch bedingte Erkrankung der Netzhaut des Auges, bei der die Lichtrezeptoren in der Netzhaut mit der Zeit absterben (degenerieren) und auf Dauer eine Erblindung die Folge ist.
Was sind denn vererbbare Degenerationen der Netzhaut ?
Es gibt zahlreiche - meist recht selten auftretende - vererbbare Degenerationserkrankungen (Erkrankungen mit zunehmender Zerstörung) der Netzhaut. Der Fachausdruck lautet “hereditäre Netzhautdystrophie”. Es handelt sich dabei um einen Defekt bzw. eine Veränderung im Erbgut, die zu einer Fehlfunktion von Teilen der Netzhaut und in der Folge, im Laufe des Lebens, zu einer teilweisen oder kompletten Zerstörung führt. Manchmal handelt es sich auch um Syndrome (Typische Kombination mehrerer Krankheiten), bei denen die Netzhautproblematik z.B. mit einer Schwerhörigkeit verknüpft ist. Allen ist gemein, daß sie auf die eine oder andere Art das Sehen einschränken oder sogar mit den Jahren zur Erblindung führen. Identifiziert wurden bisher 192 verschiedene Genregionen mit Netzhautdystrophien, wovon 144 Gene bekannt sind, d.h. man weiß welche Netzhautdystrophie dahinter steckt. Weiteres aus dieser Erkrankungsgruppe auch unter Netzhauterkrankungen.
Und was ist die Retinitis pigmentosa ?
Die bekannteste Erkrankung auf diesem Gebiet ist die Retinitis pigmentosa (kurz: RP). Es handelt sich hier jedoch nicht um eine einzelne Erkrankung mit typischem Erscheinungsbild, sondern um einen Oberbegriff für eine Gruppe von vererbbaren Netzhauterkrankungen mit zahlreichen Sonderformen, die durch Nachtblindheit, zunehmende Gesichtsfeldeinschränkung und Sehverlust charakterisiert sind.

Links normale Netzhaut, rechts Retinitis Pigmentosa mit den klassischen Pigmentierungen (2), einem wachsgelben Sehnervenkopf (3) und verengten Blutgefäßen (1) depositphotos.com
Aufgrund des Bekanntheitsgrades firmieren manche Selbsthilfegruppen für stark Sehbehinderte unter diesem Namen, obwohl auch viele Erkrankte mit ganz anders verursachten Sehbehinderungen hier Mitglied sind.
Bei der klassischen Retinitis Pigmentosa kommt es zu einem diffusen “Zerstörungsprozeß” von bestimmten Netzhautschichten. Die Lichtrezeptoren (zu Beginn die Stäbchen und später auch die Zapfen) werden zerstört. Da die Stäbchen für das Nachtsehen zuständig sind entsteht bei ihrem Untergang die typische Nachtblindheit als Früh- und Erstsymptom. Aufgrund der vom Rand her fortschreitenden Zerstörung, kommt es zu einer zunehmenden Gesichtsfeldeinschränkung, die zunächst subjektiv nicht gemerkt wird. Die jährliche Abnahe der Gesichtsfeldfläche liegt zwischen 7 und 13%. Die Nervenfasern in der Netzhaut sind bei der RP nicht betroffen. Diese teilweise erhaltene und teilweise zerstörte Netzhaut lässt sich im Querschnittsbild des OCT sehr gut darstellen. Da die “Verschaltung” der Nerven in der Netzhaut erhalten bleibt, ist es - noch im Versuchsstadium - möglich einen Mikrochip in die Netzhaut einzusetzen, der die Rezeptoren ersetzt und an die Nervenfasern angeschlossen ist (s.Netzhautimplantat). Meist tritt die Zerstörung beidseitig und seitengleich auf. Das Alter zum Zeitpunkt des Auftretens, die Geschwindigkeit und das Ausmaß des Sehverlustes, sowie das Vorhandensein von weiteren Begleiterkrankungen ist abhängig vom Vererbungsmodus und läßt sich durch die normale augenärztliche Diagnostik häufig nicht sicher voraussagen. Die gefürchtete "Endphase" mit nur noch tunnelförmig oder schlüssellochähnlichem Gesichtsfeldrest müssen glücklicherweise einige Patienten mit milder Verlaufform nicht erleben. Zur kompletten Erblindung kommt es meist erst im 5. bis 6. Lebensjahrzehnt. Diese Erkrankung gibt es übrigens auch bei Hunden (Progressive Retinaatrophie) und auch hier handelt es sich um Defekte im Erbmaterial bzw. den Chromosomen.
Wie wird sie festgestellt ?
Da sich im dritten Lebensjahrzehnt (dem Beginn der Erkrankung bei den meisten Formen) viele Menschen - mangels Beschwerden - selten einer kompletten Diagnostik bei einem Augenarzt unterziehen bzw. überhaupt dort hingehen, fällt dies zunächst nur dem Patienten selbst auf, weil er nachts immer schlechter sieht, die beginnende Nachtblindheit also als Erstsymptom. Die zunehmende Gesichtsfeldeinschränkung fällt dem Betroffenen meist lange Zeit nicht auf. Bei der Augenuntersuchung, die der Betroffenen dann meist aufgrund der Nachtsehprobleme durchführen lässt, zeigt sich dann bereits in der Gesichtsfelduntersuchung die Einschränkung und bei der Netzhautspiegelung finden sich die typischen Pigmentverklumpungen in der Netzhaut (sogenannte “Knochenkörperchen”), die der Erkrankung ihren Namen (“pigmentosa”) gegeben haben. Dies sind die dunklen Flecken rechts auf dem Netzhautbild unten aus der Bilddatenbank des Berufsverbandes der Augenärzte. Hier sind schon Netzhautteile zerstört und hier sieht der Patient schon nichts mehr. Sind im fortgeschrittenen Stadium bereits die Zapfen befallen, kommt es dazu zu erhöhter Blendungsempfindlichkeit, Farb- und Kontrastsehstörungen und schließlich zu einer Sehminderung auch im Zentrum.
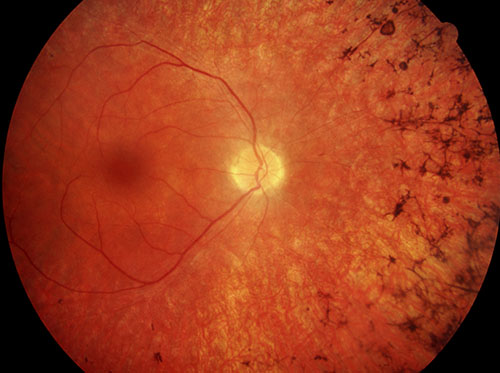
Die bei der Entdeckung entstandene und heute noch verwendete Bezeichnung als “Retinitis” ist übrigens falsch, da dies eine Netzhautentzündung wäre, die hier aber nicht vorliegt. Richtiger ist der Begriff “Retinopathia pigmentosa”, also “krankhafte Netzhautveränderung mit Pigmentierungen”, der sich aber nicht durchsetzen konnte.
Weitere Spezialuntersuchungen wie das ERG (Elektroretinogramm), bei dem die Reaktionsfähigkeit der Netzhautrezeptoren (Stäbchen und Zapfen) gemessen wird, bestätigen dann die Diagnose. Diese Untersuchung ist übrigens bereits in einem frühen Stadium auffällig.
Wie kann ich denn erfahren wie es mit meinem Sehvermögen als Betroffener und dem meiner Kinder weitergeht ?
Ob aber überhaupt ein Risiko für die Weitervererbung an die eigenen Nachkommen besteht und wie hoch dieses sein kann, darüber kann man sich heute bei entsprechend spezialisierten Fachärzten für genetische (vererbungstechnische) Fragestellungen informieren. Diese sogenannten “Humangenetiker” können mit den modernen Verfahren der Genanalyse (genaue Darstellung des menschlichen Erbgutes) inzwischen recht gut unterscheiden welche Form vorliegt und wichtige Informationen für die weitere - insbesondere auch eigene - Lebensplanung geben. Je nach betroffenem Gen kommt es nämlich zu einem gänzlich unterschiedlichem Verlauf und verschiedener Vererbungswahrscheinlichkeit.
Wie häufig kommt denn das vor ?
Mit einem allgemeinen Risiko von 1:4000 von dieser Erkrankung betroffen zu sein, handelt es sich zwar um eine seltene Erkrankung aber auch nicht so selten, daß sie nicht von Bedeutung wäre. Sie ist eine der häufigsten Ursachen für Sehbehinderung und Erblindung im mittleren Altersabschnitt. Die Häufigkeit der RP wird weltweit auf etwa 3 Millionen und in Deutschland auf 30-40 Tausend Betroffene geschätzt. Bisher sind über 45 Gene (Orte auf den Chromosomen) für die RP bekannt. Die häufigste Form mit mehr als 90 % ist die Primäre Retinitis Pigmentosa. Schätzungsweise jeder 80. Mensch trägt ein „ungünstig“ verändertes RP-Gen in sich, also eine Erbinformation, die die Entwicklung dieser Netzhauterkrankung bei Gen-Trägern oder seinen Nachkommen in Gang setzen kann.
Was für typische Verläufe gibt es denn ?
Bei der häufigsten autosomal rezessiv vererbten Form kommt es frühzeitig zu eingeschränktem Gesichtsfeld und Nachtblindheit. Das eingeschränkte Gesichtsfeld macht sich durch Ausfälle nach den Seiten bemerkbar, die dazu führen, daß man Dinge die dort passieren übersieht. Der Ausfall schreitet nach innen und außen voran und führt zum sogenannten “Flintenrohrgesichtsfeld”, man sieht wie durch ein schmales Rohr bzw. man kann zwar einen kleinen Vogel auf dem Dach genau sehen, das zugehörige Haus und den Rest des Daches aber nicht. Die Orientierung im Alltag ist für die Betroffenen stark eingeschränkt. Er stolpert oft und stößt Gegenstände um. Eine sichere Teilnahme am Strassenverkehr (auch als Fußgänger !) ist nicht mehr möglich.
Beim autosomal dominaten Vererbungsmodus tritt die Nachtblindheit erst mit im 3. Lebensjahrzehnt auf, führt aber letztendlich zur Erblindung.
Wie sind die therapeutischen Möglichkeiten ?
Trotz zahlreicher Erkenntnisse über den Hintergrund der Krankheit sind die therapeutischen Möglicheiten leider noch schlecht. Die meisten Medikamente und die Gentherapie sind noch im Tierversuchsstadium und noch nicht erhältlich. So beschränkt sich die Betreuung bisher auf die Anpassung entsprechender Brillen, vor allem mit UV-Schutz und Filtern für bestimmte Wellenlängen des Lichtes (Kantenfiltergläser) und die Anpassung von vergrößernden Sehhilfen.
Derzeit läuft allerdings eine vielversprechende Studie inwiefern eine elektrische Stimulierung der Netzhaut durch die Hornhaut (Transkorneale Elektrostimulation) hilfreich ist. Diese kann man nach Anleitung selbst zu Hause einmal pro Woche für 30 Minuten durchführen. Damit halbierte sich das Fortschreiten des Gesichtsfeldverlustes auf die Hälfte. Diese Behandlung muss lebenslang durchgeführt werden. Langfristige Ergebnisse fehlen noch und daher ist sie von den Kassen auch noch nicht anerkannt.
Sehr in den Medien sind die elektronischen Netzhautimplantate, die bei bereits erblindeten Betroffenen die abgestorbenen Photorezeptoren ersetzen sollen. Nach Erfassen der Umwelt durch eine Kamera werden die noch intakten Nerven der Netzhaut entsprechend dem gesehenen Bild gereizt und diese senden dann die Information an das Sehzentrum im Gehirn weiter, so wie sie es früher mit den Signalen aus den Photorezeptoren getan haben. Dies ist jedoch noch im Versuchsstadium und erst wenige Patienten profitieren davon und erreichen auch nur ein schemenhaftes Sehen. Verschiedene Arbeitsgruppen arbeiten technisch unterschiedlichen Lösungen. Man kann als bisher erfolgreichste Ansätze die epiretinalen Implantate (Retinastimulator wird auf die Netzhaut gesetzt) und die subretinalen Implantate (Chip unter den noch verbliebenen Netzhautschichten, anstelle der zerstörten Rezeptoren) unterscheiden.Beispiele für ersteres sind das “Argus II System”, für das in einigen Ländern schon eine Zulassung besteht und das deutsche “EPIRET3-System” das erst bei 6 Patienten eingesetzt wurde. Ein Beispiel für letzteres ist das Tübinger “Retina Implant-System” das bisher bei 35 Blinden eingesetzt wurde und als bestes Ergebnis eine Sehschärfe von 3% erreichte.
Derzeit läuft auch eine Studie an 5 Patienten bei der mittels Genfähren (Viren, die Erbmaterial in den Körper bringen) lichtempfindliche Protein-Kanäle, die aus Algen, Pilzen oder Bakterien stammen, in noch intakte Zellen der Netzhaut eingebaut werden. Diese übernehmen dann die Aufgabe der Lichtrezeptorzellen und erlauben ein zumindest teilweise wiederhergestelltes Sehen.
Internet:
Argus 2 Retinaprothesensystem:
(Stand 16.12.2024)